

Abstract
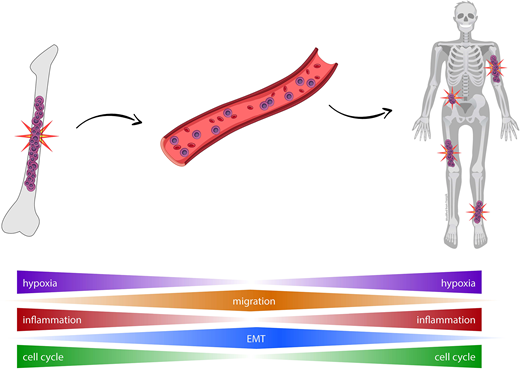
Background. The number of CTC predicts risk of transformation in smoldering MM and survival in active MM. Growing evidence suggests that as the tumor progresses and the microenvironment becomes hypoxic, clonal plasma cells (PC) constantly invade new regions of the bone marrow (BM) through induced systemic recirculation. Of note, the frequency of CTCs is typically low and thus, it is conceivable that the dissemination of MM depends on few tumor cells with unique features that induce them to egress the BM and spread the disease through peripheral blood (PB). This hypothesis has not been yet demonstrated because the transcriptional profile of CTCs in MM has not been investigated.
Aim. To identify gene regulatory networks related to MM dissemination by comparing the transcriptional profile of CTCs with patient-matched BM clonal PCs.
Methods. We used FACS to isolate CTCs and BM clonal PCs of paired PB and BM samples from 34 patients: 24 newly diagnosed MM, 9 relapsed MM and 1 MGUS. Transcriptomes were analyzed using Affymetrix arrays (n =31) and the BD WTA Precise assay was used for single-cell RNA sequencing (scRNAseq, n =3). Data was analyzed using Gene Set Enrichment Analysis (GSEA) and Limma for bulk and Seurat for scRNAseq data. The prognostic value of deregulated genes (FDR <0.1 & logFC >0.5) was investigated using a Cox-regression model in the CoMMpass dataset (n =553, IA11 release). The role of specific deregulated genes was evaluated by shRNA knockdown and blocking using a monoclonal antibody (mAb).
Results. Transcriptomic profiling of patient matched CTCs and BM clonal PCs revealed a high correlation in gene expression (r =0.93; p =10-16). Only 45 genes emerged as significantly deregulated in CTCs, and GSEA unveiled biological functions related to inflammatory and interferon response (e.g. CCL5), signaling by IL-6/JAK/STAT3, IL-2/STAT5 and TNF via NFKB (CD44), the epithelial mesenchymal transition (EMP3), mitotic spindle and G2M checkpoints (TOP2A), or E2F targets (BIRC5). A high correlation in gene expression was also observed by scRNAseq (r =0.9; p =10-16), with only 31 genes (e.g. MALAT1, B2M, RHOH, ENAM or DUSP5) differentially expressed (adj.p <0.01). Under the hypothesis that genes deregulated in CTCs could be markers of dissemination and therefore of a more aggressive disease, we evaluated their prognostic significance in CoMMpass, assuming that BM clonal PCs with higher expression of these genes would be enriched in tumor cells with CTC-features. Accordingly, the expression levels of 12/33 upregulated genes in CTCs from bulk RNAseq data were associated with significantly different progression free survival (PFS). In the multivariate analysis including the above mentioned 33 genes and the R-ISS, only FLNA retained independent prognostic value; patients showing higher FLNA expression (n =185/553) had a significantly inferior PFS vs the remaining cases (medians of 22 and 47 months, respectively; p <.001). Of note, patients with higher FLNA expression also displayed significantly higher mutational burden (p =.003). After confirming that genes deregulated in CTCs were clinically meaningful, we selected two genes upregulated in CTCs (FLNA and CD44) to investigate their functional role. Interestingly, knockdown of both genes in the U266 MM cell line significantly altered actin polymerization (p ≤.003) leading to reduce migration (2-fold, p ≤0.01). Furthermore, we evaluated the effect of an anti-CD44 mAb in CB17/Icr-PrKdcscid/Crl immunodeficient mice treated with clodronate to avoid potential phagocytosis. After inoculation of MM1S-GFP intravenously and MM1S-tomato subcutaneously in Matrigel, mice were left untreated (n =11) or treated with a weekly intravenous injection of anti-CD44 (n =11) for one month and monitored weekly by bioluminescence. Overall, there was a significant reduction in the plasmacytoma volume (5-fold, p <.001) and cell engraftment of circulating MM1S cells (42-fold, p =.003) in treated mice.
Conclusions. This is the first study analyzing the transcriptional profile of CTCs in MM. Our results reveal that gene expression of CTCs is almost identical to that of patient-matched BM clonal PCs, except for a few genes that are involved in interferon and inflammatory response, hypoxia, cell cycle and migration. Importantly, some of these genes are related to more aggressive disease and therefore, may represent novel therapeutic targets to overcome disease dissemination.
Rios:Amgen, Celgene, Janssen, and Takeda: Consultancy. Martinez-Lopez:Pfizer: Research Funding; Vivia: Honoraria; Celgene: Honoraria, Research Funding; Janssen: Honoraria, Research Funding; BMS: Research Funding; Novartis: Research Funding. Hajek:Janssen: Consultancy, Honoraria, Research Funding; Amgen: Consultancy, Honoraria, Research Funding; Celgene: Consultancy, Honoraria, Research Funding; Takeda: Consultancy, Honoraria, Research Funding; Bristol-Myers Squibb: Consultancy, Honoraria, Research Funding. San-Miguel:Sanofi: Honoraria; Celgene: Honoraria; Janssen: Honoraria; Amgen: Honoraria; Novartis: Honoraria; BMS: Honoraria; Roche: Honoraria.

